Conservation of Saola: genetic diversity, adaptation and breeding strategies
The Saola, also known as the Asian unicorn, is a critically endangered mammal found in Vietnam and Laos. It faces threats from poaching, habitat loss due to deforestation, agriculture, logging, and urban development. Genetic studies reveal a population bottleneck, emphasizing the urgency of conservation actions to protect this unique lineage from extinction. A new reference genome for the Saola species has been constructed using experimental methods such as DNA fragment assembly, quality assessment with Bioanalyzer, phylogenetic analysis, and evaluation of genetic health metrics.
Citation: The Following Is An Unpublished ‘News And Views’ Style Discussion Written By Me About “Garcia-Erill, G.; Liu, S.; Le, M. D.; Hurley, M. M.; Nguyen, H. D.; Nguyen, D. Q.; Nguyen, D. H.; Santander, C. G.; Sánchez Barreiro, F.; Gomes Martins, N. F.; Hanghøj, K.; Salleh, F. M.; Ramos-Madrigal, J.; Wang, X.; Sinding, M.-H. S.; Morales, H. E.; Stæger, F. F.; Wilkinson, N.; Meng, G.; Pečnerová, P.; Yang, C.; Rasmussen, M. S.; Schubert, M.; Dunn, R. R.; Moltke, I.; Zhang, G.; Chen, L.; Wang, W.; Cao, T. T.; Nguyen, H. M.; Siegismund, H. R.; Albrechtsen, A.; Gilbert, M. T. P. and Heller, R. (2025). Genomes of critically endangered saola are shaped by population structure and purging, Cell . Available from: https://doi.org/10.1016/j.cell.2025.03.040 ” for general audience.
Contents
- TITLE
- KEYWORDS (See section for definition 10. DEFINITION)
- SUMMARY
- INTRODUCTION
4.1 The Critical Status of the Saola: Challenges and Conservation Efforts
4.1.1 Threats to the Saola’s Survival
4.1.2 Conservation Initiatives and Challenges
4.1.3 Unique Characteristics and Genetic Concerns - 4.2 Genetic Diversity, Genetic Load, and Adaptation in Plant and Animal Breeding
- 4.2.1 The Genome and Genetic Variation
- 4.2.2 Measuring Genetic Diversity
- 4.2.3 Cross-Population Breeding and Adaptation
- 4.2.4 Outbreeding and Genetic Dilution
- 4.2.5 Balancing Genetic Diversity and Adaptation
- 4.2.6 Strategic Breeding Methods
- RESEARCH DESIGN AND EVALUATION (AKA PHILOSOPHY OF SCIENCE OR SCIENTIFIC METHOD)
5.1 Table - METHODS
6.1 Sample Source
6.2 DNA Extraction and Library Preparation
6.3 Sequencing Platforms and Data Processing
6.4 Genome Assembly and Annotation
6.5 Population Genomic Analyses
6.6 Demographic History Reconstruction
6.7 Phylogenetic and Evolutionary Analyses
6.8 Simulations and Functional Analyses
6.9 Additional Analyses - RESULTS
7.1 High-Quality Draft Genome Assembly of the Endangered Saola Species And Insights into Its Evolutionary History
7.2 Population Genomic Analysis Of Saola Northern And Southern Populations
7.3 Genome-Wide Heterozygosity and Functional Genetic Diversity Analysis in Various Animal Species: A Focus on the Saola
7.4 Genetic Diversity Analysis in Saola Populations: A Study on Nucleotide Diversity and Runs of Homozygosity (ROH)
7.5 Demographic History of Saola: An Analysis Using Pairwise Sequentially Markovian Coalescent (PSMC)
7.6 Analysis of Genetic Load Simulation And Conservation Breeding Stimulation in Saola Populations - DISCUSSION
8.1 Genetic Diversity, Genetic Load and Adaptation: Key Considerations In Conservation Outbreeding
8.1.1 Balancing Genetic Diversity, Genetic Load and Adaptation
8.1.2 Enhancing Adaptability and Resilience Due to Genetic Rescue
8.1.3 Loss of Adaptation Due to Outbreeding 8.2 Simple metrics and strategic consideration for conservation outbreeding
8.2.1. Effective Population Size (Ne)
8.2.2. FST Genetic Distance
8.2.3. Inbreeding Coefficient (F) - 8.3 Complex metrics and strategic consideration for conservation outbreeding
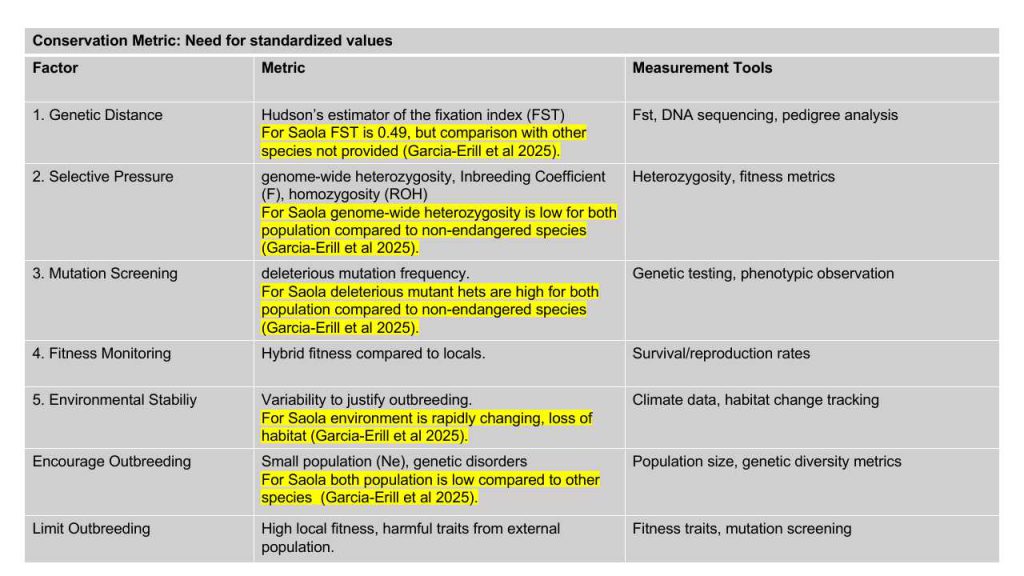
- 8.3.1 Genetic Distance
- 8.3.2 Selective Pressure
- 8.3.3 Mutation Screening
- 8.3.4 Fitness Monitoring
- 8.3.5 Environmental Stability
- 8.3.6 Breeding Strategies and Genetic Drift
- 8.4 Practical Rules for Conservation Strategic Outbreeding
- 8.4.1 Encourage Controlled Outbreeding:
- 8.4.2 Limit Outbreeding in Specific Cases:
- 8.4.3 Risk Assessment and Strategic Management:
- 8.5 Conservation: Outbreeding Decisions and Results Example
- 8.6 Proposed Workflow for In Vitro Innovation for Quick Multi-Generational Expansion of Vulnerable and Endangered Species
- 8.6.1 Field Work: Sample Collection for Scientific Research
- 8.6.2 Lab Work: Preparation of Stem Cells from Collected Samples for in vitro conservation breeding
- REFERENCES
9.1 Introduction section
9.2 Methods section
9.3 Discussion section - DEFINITION
1. TITLE
Conservation of Saola: genetic diversity, adaptation and breeding strategies.
2. KEYWORDS (See section for definition 10. DEFINITION)
Saola, ‘Asian Unicorn’, Critically Endangered, Conservation Efforts, Threats to Survival, Habitat Loss, Poaching, Genetic Diversity, Genetic Load, Inbreeding Depression, Outbreeding Depression, Gene Flow, Migration Load, Adaptation, Breeding Strategies
3. SUMMARY
Observation: The Saola, also known as the Asian unicorn, is a critically endangered mammal found in Vietnam and Laos. It faces threats from poaching, habitat loss due to deforestation, agriculture, logging, and urban development. Genetic studies reveal a population bottleneck, emphasizing the urgency of conservation actions to protect this unique lineage from extinction. Preserving the Saola is crucial for maintaining global biodiversity and requires immediate and ongoing efforts from conservationists, scientists, governments, and local communities. Aim: The aim of the study was to construct and evaluate a new reference genome for the endangered Saola species. The genome was useful to evaluate the genetic health of two Saola populations using different metrics and determine if mixing the two populations can restore both their numbers and genetic health. Experimental Methods: Experimental Methods were used, including DNA fragment assembly, quality assessment with Bioanalyzer, phylogenetic analysis, evaluation of genetic health metrics such as genome-wide heterozygosity, Runs Of Homozygosity (ROH), Hudson’s estimator of the fixation index (FST), and demographic history. Results: The study resulted in a high-quality genome assembly that is suitable for various genomic analyses at gene level (95.2% complete for gene content) and chromosome level (90% complete chromosome organization). However, it may be limited for comparison between two Saola populations as only the northern population was used to construct the reference genome instead of also using the southern population to construct a net pan-genome. The phylogenetic tree constructed revealed a close relation to the Water Buffalo and Cattle, with a divergence occurring approximately 20 million years ago during the Neogene Miocene epoch. The Saola FST was high at 0.49, and compared to other animals, its genome-wide heterozygosity was low and bad mutations or potentially bad mutations in hets were high. The nucleotidy diversity and ROH analysis showed that both populations had comparable values, and their demographic analysis revealed a steep drop of population size from 10,000 to 5000 individuals during the Last Glacial Maximum (LGM) or 20,000 years ago in timescale. Inference and Future Directions: The study offers valuable insights into the evolutionary history of the Saola and provides a foundation for future conservation strategies. The importance of using multiple methods and statistical validation is emphasized, as well as the potential value of comparative demographic studies to enhance understanding. Future studies should incorporate both populations in constructing de novo reference genome, provide a measure of significance such as classical Confidence Intervals, T-tests p-values and/or modern bootstrap p-values, compare other metrics such as ROH analysis (homozygosity) and nucleotide diversity. Though stimulation of northern and southern breeding did not show any statistical significant improvement in genetic health, practically as the population of Saola are vanishingly low, breeding the two populations is the last resort to save them.
4. INTRODUCTION

The saola, also known as the Asian unicorn, is a critically endangered mammal found in Vietnam and Laos. It faces threats from poaching, habitat loss due to deforestation, agriculture, logging, and urban development. Conservation efforts include establishing protected areas and anti-poaching measures, but challenges such as remote monitoring and capturing wild saolas for breeding programs persist. Genetic studies reveal a population bottleneck, emphasizing the urgency of conservation actions to protect this unique lineage from extinction. The intricate interplay between genetics, evolution, and breeding strategies underpins our understanding of life’s complexities. This knowledge allows us to navigate the delicate balance between genetic diversity and adaptation for the betterment of both biodiversity and human endeavors. Preserving the saola is crucial for maintaining global biodiversity, requiring immediate and ongoing efforts from conservationists, governments, and local communities.
4.1 The Critical Status of the Saola: Challenges and Conservation Efforts
The saola, often referred to as the “Asian Unicorn,” holds a unique and critical position in the animal kingdom. This elusive creature is not only one of the rarest mammals on Earth but also a symbol of the urgent conservation challenges facing many species today. Since its discovery in 1992, the saola has been a subject of fascination and concern, as it now faces a grim future. With fewer than 100 individuals believed to remain in the wild and no confirmed sightings since 2013, the fears surrounding its extinction are mounting. The saola is officially listed as Critically Endangered on the IUCN Red List, highlighting the pressing need for immediate and effective conservation efforts.
4.1.1 Threats to the Saola’s Survival
The survival of the saola is jeopardized by several significant threats. The most pressing of these is indiscriminate poaching, where poachers utilize wire snares primarily intended for other bushmeat species. This not only directly endangers the saola but also disrupts the delicate balance of its ecosystem. Additionally, habitat loss presents a substantial risk, primarily driven by deforestation, agriculture, logging, and urban development. These activities have severely impacted the saola’s habitat in the Annamite Mountains, straddling the borders of Vietnam and Laos. Compounding these issues is the saola’s slow reproductive rate and small population size, which heighten the risk of inbreeding depression—a phenomenon that can threaten the long-term viability of its genetic diversity.
4.1.2 Conservation Initiatives and Challenges
Amid these challenges, various conservation initiatives are underway to protect the saola and its habitat. Efforts include the establishment of protected areas and the enforcement of anti-poaching measures. However, effective enforcement is complicated by the region’s remoteness, which makes monitoring and regulating snaring practices exceedingly difficult. Additionally, attempts to locate and capture wild saola for breeding programs are thwarted by the species’ elusive nature and the challenging terrain of its habitat. To enhance conservation efforts, groups are employing innovative techniques such as camera traps, environmental DNA analysis, and engaging with local communities to raise awareness and involve them in preservation activities.
4.1.3 Unique Characteristics and Genetic Concerns
The saola is uniquely adapted to thrive in dense, wet, evergreen forests at specific elevations within the Annamite Mountain Range. As the only known member of the Pseudoryx genus, the saola occupies a distinct evolutionary niche that underscores its importance in the biodiversity of the region. However, recent genetic studies reveal a concerning population bottleneck, linked to drastic population declines, which further complicates its chances of survival. This genetic vulnerability emphasizes the urgency of conservation actions to protect this unique lineage from irreversible loss. Preserving the saola is not just about saving a single species; it is vital for maintaining global biodiversity. As such, it requires immediate and ongoing efforts from conservationists, governments, and local communities to prevent its extinction in the wild. By addressing these critical issues and fostering collaborative conservation strategies, there is still hope for the saola to thrive in its natural habitat. The time to act is now, as each passing moment brings this remarkable creature closer to the brink of extinction.
4.2 Genetic Diversity, Genetic Load, and Adaptation in Plant and Animal Breeding
In plant and animal breeding, particularly within conservation efforts aimed at preserving biodiversity, three fundamental aspects demand careful consideration: genetic diversity, genetic load, and adaptation. The intricate interplay between genetics, evolutionary biology, and breeding strategies forms the cornerstone of our understanding of life’s complex web. This interplay delves into the very heart of an organism’s genetic makeup, its ability to adapt to changing environments, and the impact of selective breeding practices on genetic diversity and trait expression.
4.2.1 The Genome and Genetic Variation
At the core of this intricate tapestry lies the genome—a comprehensive blueprint composed primarily of DNA and RNA components, serving as the ultimate guide for an organism’s development, function, and reproduction. The origin of distinct traits stems from the dynamic interplay between an organism’s genotype and its surroundings. Genetic variation, a cornerstone of evolutionary biology, is defined by alleles—alternate forms of genes present in related individuals. Homozygotes, with identical alleles (e.g., AA or bb), contrast with heterozygotes who exhibit distinct allelic variations (e.g., Ab or Bb).
4.2.2 Measuring Genetic Diversity
Measuring genetic diversity involves analyzing nucleotide variation, which can significantly influence gene expression, ultimately affecting traits and an organism’s fitness for survival. Heterozygosity reflects the shared alleles between individuals, while structural variations such as inversions, translocations, fragile sites, point mutations, mitochondrial DNA changes, and epigenetic modifications further contribute to trait variation and fitness levels.
4.2.3 Cross-Population Breeding and Adaptation
The relationship between genetic diversity and environmental adaptation is a key area of investigation through breeding strategies. Cross-population breeding aims to enhance desirable traits by blending alleles from different populations. While this approach may boost growth and fertility, it also carries risks of disrupting unique local adaptations. This delicate balance between harnessing genetic diversity and preserving adaptation introduces ethical, cultural, and biological complexities that require strategic considerations.
4.2.4 Outbreeding and Genetic Dilution
Outbreeding can bring about beneficial genes but may also erode local adaptations, raising questions about how external influences impact population health and survival. The loss of unique adaptation traits occurs when subpopulations interbreed with external groups, leading to genetic dilution. Therefore, preserving biodiversity and ensuring long-term survival demands an understanding of these dynamics for effective conservation efforts.
4.2.5 Balancing Genetic Diversity and Adaptation
Achieving a delicate balance between maintaining genetic diversity and adapting to the environment involves managing gene flow through controlled exchange or maintaining large populations that minimize genetic drift while preserving beneficial alleles. Advanced technologies like next-generation sequencing and genotyping arrays provide unprecedented insights, enabling comprehensive data analysis. Theoretical models further enhance our ability to predict outcomes and make informed decisions.
4.2.6 Strategic Breeding Methods
Strategic breeding methods are essential for ensuring resilient, healthy populations well-adapted to their environments. By carefully considering gene flow mechanisms and avoiding inbreeding or outbreeding, scientists can optimize genetic health and adaptation potential. This fosters the creation of thriving populations capable of prospering within diverse ecosystems.
5. RESEARCH DESIGN AND EVALUATION (AKA PHILOSOPHY OF SCIENCE OR SCIENTIFIC METHOD)
5.1 Table
| Observation | The Saola, also known as the Asian unicorn, is a critically endangered mammal found in Vietnam and Laos. It faces threats from poaching, habitat loss due to deforestation, agriculture, logging, and urban development. Genetic studies reveal a population bottleneck, emphasizing the urgency of conservation actions to protect this unique lineage from extinction. Preserving the Saola is crucial for maintaining global biodiversity and requires immediate and ongoing efforts from conservationists, scientists, governments, and local communities. |
| Hypothesis | Breeding of the two Saola populations will restore their genetic health and numbers. |
| Experiment Aim | The aim of the study was to construct and evaluate a new reference genome for the endangered Saola species. The genome was useful to evaluate the genetic health of two Saola populations using different metrics and evaluate if mixing the two populations can restore both their numbers, and genetic health. |
| Experiment Method | The authors initiated the genome assembly process by utilizing DNA fragments of varying lengths. They assessed the quality and integrity of these fragments using a Bioanalyzer, which played a crucial role in selecting the optimal DNA sample for constructing a draft de novo reference genome assessed with BUSCO and PCF constructed by RACA for completeness. Phylogenetic analysis was conducted to determine the evolutionary relationship between the Saola and other species. Genetic health of the two Saola populations, northern and southern, was evaluated using metrics such as genome-wide heterozygosity, Runs Of Homozygosity (ROH), Hudson’s estimator of the fixation index (FST), and demographic history. To test if breeding between the two populations can restore Saola numbers, and genetic health, breeding was stimulated. |
| Result | The study resulted in a high-quality genome assembly that is suitable for various genomic analyses, at gene level (95.2% complete for gene content) and chromosome level (90% complete chromosome organization). However, it may be limited for comparison between two Saola populations as only northern population was used for constructing the reference genome instead of also using southern population to construct a net pan-genome. The phylogenetic tree constructed revealed a close relation to the Water Buffalo and Cattle, with a divergence occurring approximately 20 million years ago during the Neogene Miocene epoch, and this topology of the tree was supported by 0.7 fraction of blocks with topology. Saola FST was high at 0.49, and compared to other animals, its genome-wide heterozygosity was low and bad mutations or potentially bad mutations in hets was high. Saola nucleotidy diversity and ROH was comparable between both populations, and both their demographic analysis showed a steep drop of population size from 10,000 to 5000 individuals, during Last Glacial Maximum (LGM) or 20,000 years ago in timescale. Before LGM both populations shared same ancestry. Stimulations further showed that a sharp drop in population size, coincides with purging of recessive mutations from the population which has a plateauing effect on rise of genetic load. |
| Inference and Future Direction | The study offers valuable insights into the evolutionary history of the Saola, providing a foundation for future conservation strategies. The importance of using multiple methods and statistical validation is emphasized, as well as the potential value of comparative demographic studies to enhance understanding. Future studies should incorporate both populations in construction of de novo reference genome, provide a measure of significance such as classical Confidence Intervals, T-tests p-values and/or modern bootstrap p-values. Just as estimates of genome-wide heterozygosity and mutation hets were compared between Saola and other animals, for other metrics such as ROH analysis (homozygosity) and nucleotide diversity. Addressing these missed opportunities will help gain a more comprehensive understanding of the Saola’s genetics and evolutionary trajectory, ultimately aiding in the conservation of this unique and endangered species. Though stimutation of northern and southern breeding did not show any statistical significant improvement in genetic health, practically as the population of Saola are vanishingly low, breeding the two populations is the last resort to save them. |
| Hypothesis true or false? | False, because stimulated breeding showed no statistical significant restoration of genetic health. (However as Saola are so few in number, to save the species, breeding them is the only choice to give Saola a last chance for survival). |
6. METHODS
The research utilized a thorough, multi-step strategy to dissect the genetic diversity and evolutionary trajectory of this population through whole-genome sequencing. By integrating cutting-edge genomic techniques with sophisticated bioinformatics tools and robust statistical analyses, we achieved an intricate portrayal of its genetic variation, structure, and lineage—insights crucial for devising effective conservation measures tailored to the population’s unique needs.
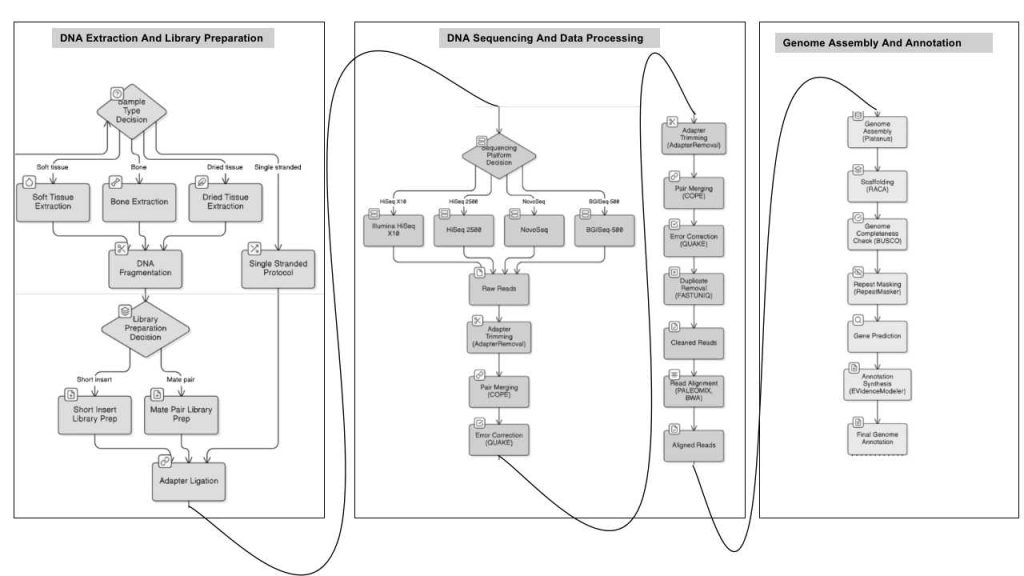
6 .1 Sample Source
The study primarily used samples collected during the initial discovery and description of the saola, which were stored at the University of Copenhagen. Additional samples were collected by the Central Institute for Natural Resources and Environmental Studies in Vietnam, consisting mainly of degraded skin, hair, or bone from trophies of indigenous hunters, and preserved tissue samples.
6.2 DNA Extraction and Library Preparation
DNA was isolated from various sample types—soft tissue, bone, and dried tissue—using the Qiagen DNeasy kit with sample-specific modifications. Bone samples underwent a specialized protocol involving bleach, EDTA, urea, and proteinase K, followed by MinElute PCR purification. Dried tissues were washed with bleach, ethanol, and water before digestion and phenol-chloroform extraction. Extracted DNA was fragmented using a Bioruptor, and size selection targeted 250 bp and 500 bp inserts for short libraries, or 2 kb and 5 kb for mate-pair libraries. For three samples, the Santa Cruz Single Stranded protocol was used, converting double-stranded DNA into single strands via Single Stranded Binding proteins, then ligating adapters with T4 DNA Ligase and T4 Polynucleotide Kinase.
6.3 Sequencing Platforms and Data Processing
Multiple platforms—Illumina HiSeq X10, HiSeq 2500, NovoSeq, and BGISeq-500—generated sequencing data, supporting both de novo genome assembly and sample sequencing. Raw reads were processed with AdapterRemoval for adapter trimming, COPE for merging overlapping pairs, QUAKE for error correction, and FASTUNIQ for duplicate removal. Cleaned reads were aligned to the reference genome using PALEOMIX and BWA.
6.4 Genome Assembly and Annotation
The genome was assembled with Platanus, utilizing short-insert libraries, and scaffolded with RACA using paired-end and comparative data. Genome completeness was assessed with BUSCO, and transposable elements were masked using RepeatMasker. Gene prediction combined homology-based and de novo methods, with EVidenceModeler synthesizing results into a final annotation.
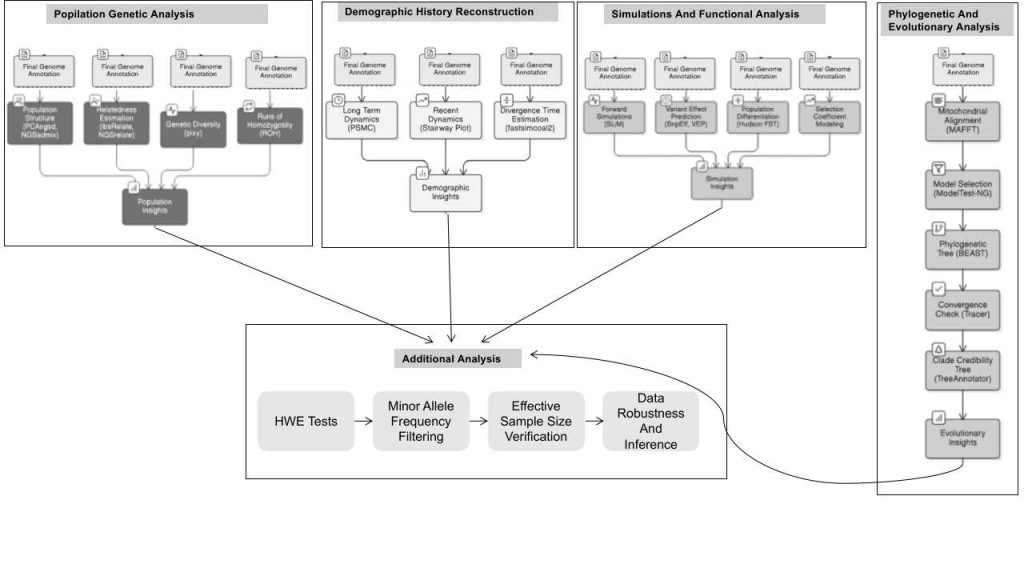
6.5 Population Genomic Analyses
Tools such as PCAngsd and NGSadmix analyzed population structure and admixture. Relatedness was estimated with ibsRelate and NGSrelate, while genetic diversity metrics were obtained via pixy. Runs of homozygosity (ROH) were examined to infer inbreeding levels. These analyses provided insights into population structure, diversity, and relatedness.
6.6 Demographic History Reconstruction
Long-term and recent population dynamics were inferred with PSMC and Stairway Plot, respectively. Divergence times between populations were estimated using fastsimcoal2. These models elucidated historical population size changes and splits.
6.7 Phylogenetic and Evolutionary Analyses
Mitochondrial genomes were aligned with MAFFT, with substitution models selected via ModelTest-NG, and phylogenetic trees reconstructed using BEAST. Convergence was verified with Tracer, and maximum clade credibility trees were generated with TreeAnnotator.
6.8 Simulations and Functional Analyses
Forward simulations in SLiM modeled genetic load accumulation and assessed conservation management strategies under various scenarios. Variant effects were predicted with SnpEff and VEP. Population differentiation was quantified with Hudson’s FST, and selection coefficients were incorporated into simulations.
6.9 Additional Analyses
The study employed multiple statistical and bioinformatics approaches, including HWE tests, minor allele frequency filtering, and effective sample size verification, to ensure data robustness and accurate inference.
7. RESULTS
7.1 High-Quality Draft Genome Assembly of the Endangered Saola Species And Insights into Its Evolutionary History
(Figure 1 of paper Garcia-Erill 2025 et al)
(Figure S1 of paper Garcia-Erill 2025 et al)
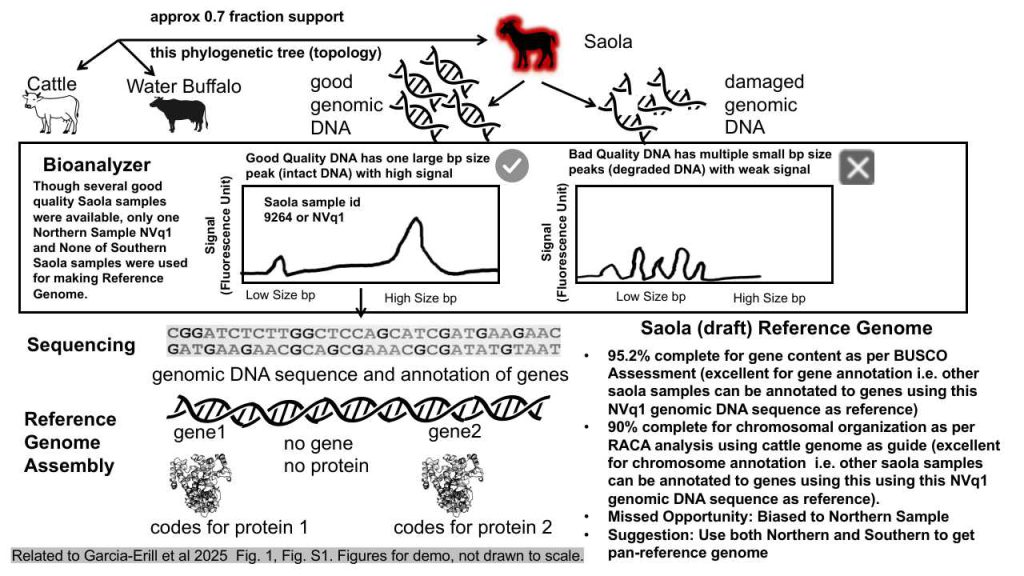
Introduction: The Saola, a critically endangered species indigenous to Southeast Asia, is the subject of a comprehensive genomic study aimed at constructing and evaluating a new reference genome. This study employs a multitude of advanced methods, including Bioanalyzer, Repeat-Enabled Alignment to Consensus (RAC), Benchmarking Universal Single-Copy Orthologs (BUSCO), and phylogenetic analysis, to ensure the quality and accuracy of the Saola genome assembly. The overarching goal is to provide a robust genetic framework that can support future conservation efforts and comparative genetic studies.
Experimental Methods: The authors initiated the genome assembly process by utilizing DNA fragments of varying lengths. The quality and integrity of these fragments were meticulously assessed using a Bioanalyzer, which played a crucial role in selecting the optimal DNA sample for the construction of a draft de novo reference genome. The selected sample, identified as NVq1, exhibited a Bioanalyzer plot with a prominent peak at 14,400 bp and a smaller peak at 100 bp, the latter potentially attributed to degradation or experimental artifacts. The genome’s completeness was evaluated using BUSCO, which indicated a 95.2% completeness for gene content, signifying an excellent reference for gene annotation. Furthermore, the chromosomal organization was assessed using RAC, guided by the well-studied cattle genome, resulting in a 90% completeness, which is commendable for chromosome annotation. The N50 value of the Saola genome was determined to be 79 Mb, reflecting the genome’s complexity and the challenges associated with assembling less-studied genomes compared to more extensively researched ones, such as the cattle genome with an N50 of 103 Mb. Phylogenetic analysis, conducted using sliding windows across the Saola genome, revealed varying phylogenetic placements, and Bayesian inference was employed to estimate divergence times with a 95% high posterior density, providing a probabilistic framework for understanding the evolutionary timeline.
Results and Inference: The results underscore the Bioanalyzer’s importance in assessing the quality and size distribution of genomic DNA fragments prior to next-generation sequencing. The high-quality genome assembly achieved in this study is suitable for a wide range of genomic analyses, including studying synteny, tracking chromosomal rearrangements, analyzing gene clusters, and comparing chromosome evolution. However, it may be limited in studies requiring complete centromere analysis, full telomere-to-telomere assembly, and complex structural variants. The phylogenetic tree constructed to determine the evolutionary relationship between the Saola and other species indicates a close relation to the Water Buffalo and Cattle, with a divergence occurring approximately 20 million years ago during the Neogene Miocene epoch. The topology of this phylogenetic tree is supported by 0.7 fraction of blocks, providing a strong foundation for understanding the Saola’s evolutionary history.
Missed Opportunities: Despite the study’s comprehensive approach, several opportunities were missed that could have enhanced the genome assembly’s robustness and unbiased nature. Primarily, the authors focused solely on a northern population sample, neglecting the southern population. This approach introduces biases, such as allele frequency bias, structural variation bias, mapping bias, and ancestral state bias, which can affect the accuracy of genetic variation comparisons between the populations. Incorporating a pan-genome approach, using multiple references, and performing de novo assemblies for both populations could have provided a more balanced and representative genome assembly. Additionally, the study did not provide bootstrap support values for the phylogenetic tree, which are essential for assessing the reliability of the inferred evolutionary relationships. Bootstrap values from an ASTRAL analysis would have offered insights into the consistency and robustness of the phylogenetic inferences, highlighting areas that may require further investigation or additional data.
Conclusion: In conclusion, this study offers valuable insights into the Saola’s evolutionary history, providing a foundation for conservation efforts and comparative genetic studies. The results emphasize the importance of using multiple references, performing de novo assemblies, and employing sensitive mapping and statistical corrections to enhance the accuracy and representativeness of genomic analyses. By addressing the missed opportunities, future studies can build upon this work to gain a more comprehensive understanding of the Saola’s genetics and evolutionary trajectory, ultimately aiding in the preservation of this unique and endangered species.
7.2 Population Genomic Analysis Of Saola Northern And Southern Populations
(Figure 2 of paper Garcia-Erill 2025 et al)
(Figure S2 of paper Garcia-Erill 2025 et al)
(Figure S3 of paper Garcia-Erill 2025 et al)
(Figure S4 of paper Garcia-Erill 2025 et al)

Introduction: The Saola, a critically endangered species indigenous to Vietnam and Laos, is divided into two distinct populations: the northern Saola and the southern Saola. To elucidate the population genomic structure and diversity of this elusive species, researchers employed a comprehensive suite of analytical methods. These included Principal Component Analysis (PCA), admixture proportions, Hudson’s estimator of the fixation index (FST), runs of homozygosity (ROH), and heterozygosity estimates. The study encompassed twenty-six Saola individuals, with fifteen from the northern population and eleven from the southern population, providing a robust dataset for analysis.
Methodology: Sequencing depth, defined as the average number of times a nucleotide is read during sequencing, is crucial for ensuring the accuracy and completeness of genetic data. A sequencing depth greater than 6X was employed in this study to guarantee high-quality data for the Saola individuals analyzed. Additionally, an error rate for transversion mutations below 0.001 was maintained to filter out potential sequencing errors. Transversion mutations, which involve the replacement of a purine with a pyrimidine or vice versa, are rarer and more likely to alter protein sequences, potentially leading to deleterious effects. By maintaining a low transversion rate, the study minimized errors and biases, ensuring that the genetic data accurately reflected the biological reality of the Saola populations.
Results: Admixture analysis revealed low levels of genetic mixing between the northern and southern Saola populations, with an FST value of 0.49 indicating significant genetic differentiation. This suggests limited gene flow between the two populations, although some individual correlations hint at familial relationships. PCA and mitochondrial DNA clustering further supported this finding by distinguishing genetic profiles unique to each group. Runs of Homozygosity (ROH) ranged from 0.2 to 0.4 fraction of the genome, although the variability remains uncertain due to the lack of standard deviation data. The kinship matrix highlighted relatedness and inbreeding within both the northern and southern populations. Heterozygosity estimates for the Saola ranged from 0.0003 to 0.0005, with the southern population exhibiting lower heterozygosity than the northern population when ROH was excluded, indicating reduced genetic diversity.
Inference: The study offers valuable insights into the genetic health and structure of the Saola population. Mitochondrial DNA analysis, PCA, and low admixture levels suggest distinct populations with limited gene flow and relatedness. Heterozygosity estimates indicate reduced genetic diversity, particularly in the southern population. The kinship matrix revealed relatedness and inbreeding within both populations, underscoring the need for conservation efforts to maintain genetic diversity. These findings are crucial for understanding the homozygosity and heterozygosity dynamics within Saola populations, highlighting the urgency of conservation measures to preserve their genetic integrity.
Missed Opportunities: Several analytical gaps were identified that could have enhanced the study’s conclusions. Statistical testing for measures such as FROH (the proportion of the genome in runs of homozygosity) and NROH (the number of runs of homozygosity) was not performed. This omission could have provided deeper insights into autozygosity and recent inbreeding levels. Furthermore, while ROH was included in heterozygosity calculations, statistical significance for comparisons between the two populations was not established using tests such as T-test or ANOVA. Addressing these gaps could have strengthened the robustness of the conclusions regarding the genetic diversity and structure of the Saola population.
7.3 Genome-Wide Heterozygosity and Functional Genetic Diversity Analysis in Various Animal Species: A Focus on the Saola
(Figure 3 of paper Garcia-Erill 2025 et al)
(Figure S5 of paper Garcia-Erill 2025 et al)
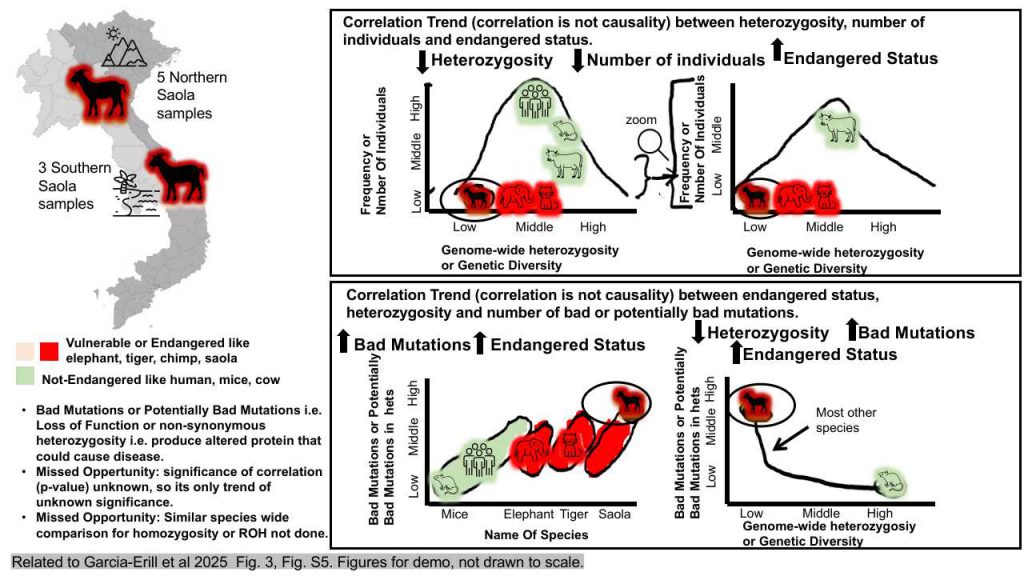
Introduction: This study investigates genome-wide heterozygosity and functional genetic diversity across multiple animal species, with a particular emphasis on the critically endangered Saola. The authors aim to compare various aspects of genetic variability among different taxa and conservation statuses to better understand how these factors influence endangered status, genetic health, and overall survival.
Experiment: The research utilized genomic data from a range of animal species, categorized by taxonomy (including animals, mammals, and bovids) and conservation status, spanning from Least Concern to Critically Endangered. The authors analyzed heterozygosity levels across these groups, employing scatter plots to visualize the data and calculating the ratio of potentially deleterious genetic variations. These variations include harmful mutations, such as loss-of-function mutations and non-synonymous heterozygosity, which can lead to the production of altered proteins that may cause disease.
Results: In alignment with their endangered status, the Saola and other endangered species exhibited significantly lower population levels compared to non-threatened species, such as humans, cattle, and mice. The findings indicated that the genome-wide heterozygosity levels of the Saola were comparable to those of other critically endangered non-bovid species, including the Tasmanian devil, snow leopard, and cheetah, as well as geographically localized bovid species like the Nilgiri Tahr and Hirola. Furthermore, the analysis revealed that endangered species, including the Saola, displayed higher levels of deleterious genetic variation compared to other mammals.
Inference: The results suggest a correlation between conservation status and genetic health among these species. The authors emphasize the importance of preserving genetic health to ensure the survival of endangered species, as the loss or reduction of essential proteins can significantly impact an organism’s health and development.
Missed Opportunity: One limitation of the study is the absence of p-values for the significance of the correlations, leaving the statistical significance of the findings uncertain. Additionally, while the study compared heterozygosity levels across different taxa and conservation statuses, it did not examine homozygosity rates of runs of homozygosity (ROH) across various species. Such comparisons could provide further insights into the genetic health of the Saola in relation to other species.
Conclusion: This research underscores the urgent need for ongoing conservation efforts to protect endangered species and maintain genetic diversity across various mammal populations. The study highlights that preserving genetic health is vital for ensuring the survival of endangered species, as the loss or reduction of key proteins can have profound effects on an organism’s health and development. It is important to note that correlation does not imply causation; thus, the relationships between mutations, homozygosity, heterozygosity, and population numbers warrant further investigation through modeling, animal studies, and laboratory experiments.
7.4 Genetic Diversity Analysis in Saola Populations: A Study on Nucleotide Diversity and Runs of Homozygosity (ROH)
(Figure 4 of paper Garcia-Erill 2025 et al)
(Figure S6 of paper Garcia-Erill 2025 et al)
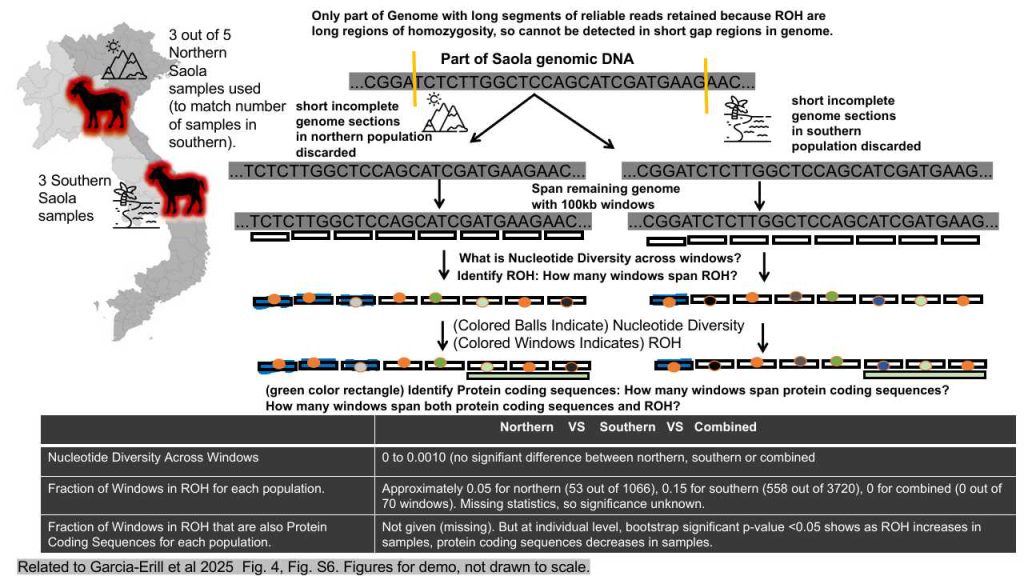
Introduction: The genetic diversity and runs of homozygosity (ROH) in Saola (Pseudoryx nghetinhensis) populations were examined to assess evolutionary history, genetic structure, and conservation potential. This study focused on three distinct populations—Northern, Southern, and a Combined group (jointly analyzed, not an actual breeding population)—to evaluate genetic diversity loss. Key metrics included nucleotide diversity, haplotype proportions, genome-wide coding sequence proportions, and single nucleotide polymorphisms (SNPs) within ROH regions. Given that longer genomic sequences enhance ROH detection, segments shorter than 10 Mb were excluded, resulting in varying genome sizes for each population. The remaining genome was analyzed using 100 kb windows, categorized as either ROH or non-ROH regions.
Results: In 100 kb windows where Tajima’s π indicated depleted genetic diversity, over 50% (and in some cases 100%) of samples exhibited ROH. This aligns with the observation that regions with reduced genetic diversity had a high prevalence of ROH. A comparative boxplot of nucleotide diversity across predicted depleted regions revealed no statistically significant differences between Northern, Southern, and Combined populations, suggesting uniform genetic diversity loss.
The study further quantified haplotype distributions by assessing 100 kb windows where:
- The same haplotype occurred in Southern samples
- The same haplotype occurred in Northern samples
- The same haplotype occurred in both populations
- The same haplotype occurred in the Combined sample
While mean values were plotted without standard deviations (limiting quantitative reliability), the analysis of ROH frequency across samples still offered insights into genetic diversity and population structure. Additionally, in regions of depleted genetic variation, increasing ROH prevalence correlated with a decline in protein-coding sequences (statistically significant, p < 0.05). However, no significant inter-population differences were detected.
Inference: The findings suggest that Saola populations may be experiencing genetic diversity loss due to widespread ROH. This has critical implications for species survival, adaptability, and breeding program viability. Further research is needed to elucidate the drivers of this decline and to inform conservation strategies.
Missed Opportunities: The study could have been strengthened by including:
- Detailed population size and distribution data to contextualize results
- The proportion of ROH windows overlapping protein-coding sequences
- Expanded genetic marker analysis (e.g., SNPs, mitochondrial DNA) for a more comprehensive assessment of diversity loss
These additions would enhance the study’s interpretative power and conservation applicability.
7.5 Demographic History of Saola: An Analysis Using Pairwise Sequentially Markovian Coalescent (PSMC)
(Figure S7 of paper Garcia-Erill 2025 et al)
(Figure 5 of paper Garcia-Erill 2025 et al)
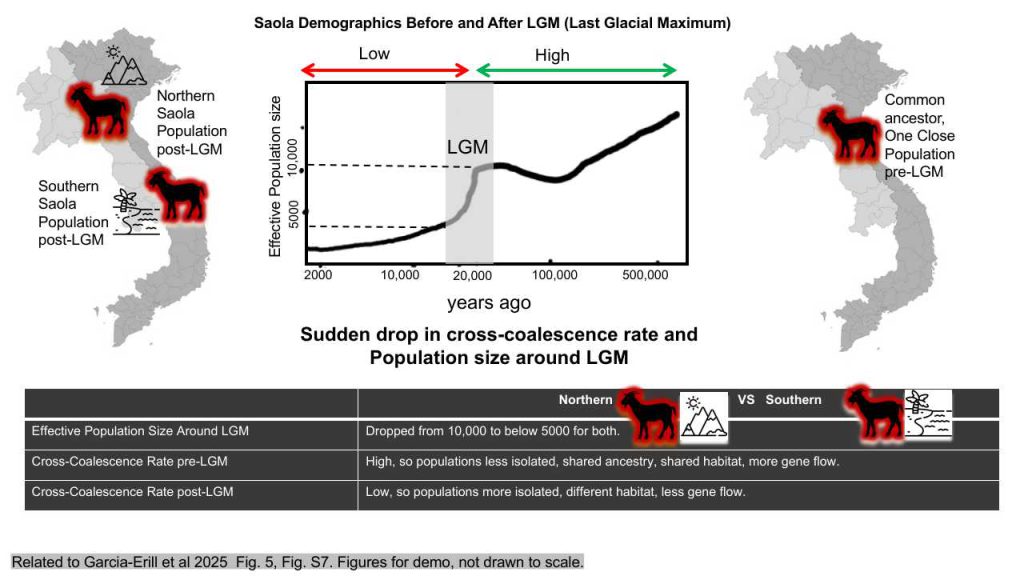
Introduction: Demographic history encompasses the historical changes in population size, structure, migration patterns, and genetic exchange that have influenced a species’ evolutionary trajectory over time. These demographic events include population expansions or contractions, founding events, population bottlenecks, migrations, admixture, and selective sweeps. Understanding these processes is crucial for comprehending the evolutionary dynamics and genetic diversity of a species.
Methods: To study the demographic history of the Saola, researchers often employ computational methods such as the Pairwise Sequentially Markovian Coalescent (PSMC). PSMC is a statistical approach that infers demographic changes in populations from whole-genome sequence data. This method models the coalescent process, which involves backtracking the common ancestors of DNA sequences from two Saola populations through time to their most recent common ancestor. By analyzing patterns of genetic variation across the genome, PSMC provides insights into past population size fluctuations and other demographic events that have shaped the evolutionary history of the Saola species.
Resultsand Inference: Using PSMC, researchers estimated the effective population sizes for two Saola populations, Northern and Southern, over time, both before and after the Last Glacial Maximum (LGM). The effective population size refers to the average number of individuals in a population that contribute to genetic variation. The results indicated that around the LGM, the effective population size for both Northern and Southern Saola dropped below 5,000 individuals. This suggests that both populations experienced a significant bottleneck during this period. The cross-coalescence rate, a measure of how closely related two populations are in terms of their genetic makeup, revealed significant findings. A high relative cross-coalescence rate in the pre-LGM period suggests substantial gene flow or shared ancestry between the populations at that time. This could indicate periods of migration, admixture, or shared habitat. Conversely, a low relative cross-coalescence rate in the post-LGM period suggests that the populations were more isolated or divergent during that time, potentially due to geographic barriers, ecological differences, or other factors that reduced gene flow.
Missed Opportunities for Further Analysis:
- While the PSMC method provides valuable insights into the demographic history of Saola populations, there are several missed opportunities for further analysis that could enhance the study’s robustness and depth.
- Statistical Validation: One significant area for further analysis involves the use of statistical validation methods to strengthen the findings. For instance, comparisons between the two populations could be significantly inferred with the application of confidence intervals (CI), likelihood ratio tests, adapting bootstrap methods to give p-values, and/or permutation tests. Confidence intervals around the estimates of effective population size or cross-coalescence rates at the LGM could help determine whether the observed patterns are robust or if they could be due to random variation. For example, if the confidence intervals for effective population size or cross-coalescence rates do not overlap between the two populations at a given time point of interest (such as the LGM), this would suggest a statistically significant difference in their demographic histories. However, since these statistics are not provided, the comparison remains inconclusive.
- Comparative Demographic Studies: Another missed opportunity lies in comparing the demographic histories of the Saola with other closely related species, such as cattle or water buffalo. Such comparative studies could provide further insights into how similar demographic events have shaped the evolutionary trajectories of these species. By examining the demographic histories of related species, researchers could identify common patterns and unique differences, thereby offering a broader understanding of the evolutionary processes at play.
Conclusion: In conclusion, the PSMC method offers valuable insights into the demographic history of Saola populations over time. The results demonstrate that both Northern and Southern Saola populations encountered significant bottlenecks during the LGM period, which affected their effective population size and cross-coalescence rate. Despite the missed opportunities for further analysis, this study underscores the importance of using computational methods like PSMC to understand the complex demographic events that have shaped the evolutionary trajectories of species over time. Future research could build on these findings by incorporating statistical validation methods and comparative demographic studies to provide a more comprehensive understanding of the Saola’s evolutionary history.
7.6 Analysis of Genetic Load Simulation And Conservation Breeding Stimulation in Saola Populations
(Figure 6 of paper Garcia-Erill 2025 et al)
(Figure S8 of paper Garcia-Erill 2025 et al)
(Figure S9 of paper Garcia-Erill 2025 et al)
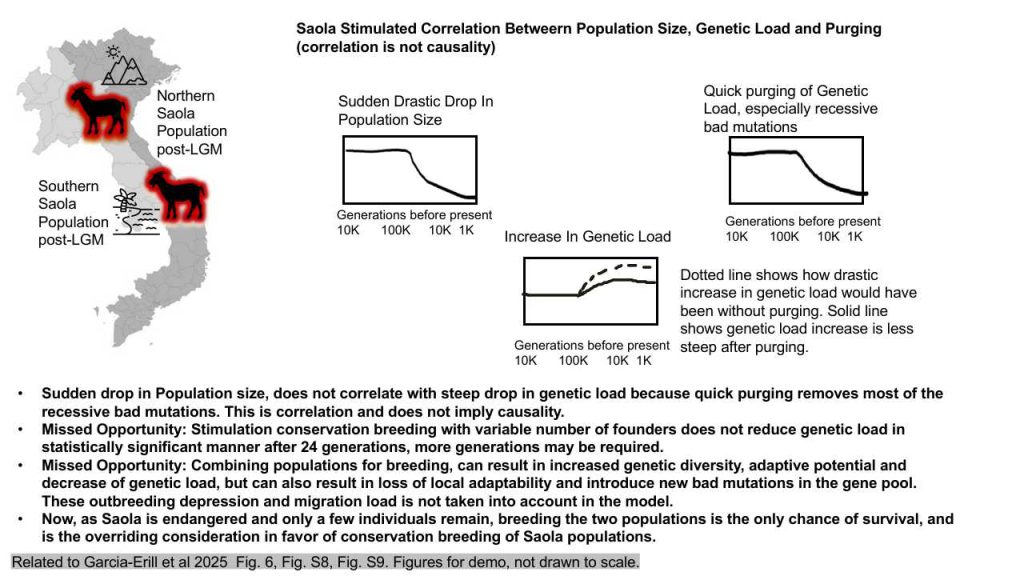
Introduction: This analysis presents a thorough investigation into the simulation of genetic load within the critically endangered Saola species. Utilizing SLiM software, forward simulations were conducted to replicate the population’s demographic history. The primary focus was on the relationship between a sharp population decline, the accumulation of deleterious mutations, the purging of genetic load, and the net realized genetic load following this purging process.
Methods: The SLiM software was employed to perform forward simulations that mimic the demographic history of the Saola population. These simulations were designed to explore the dynamics of genetic load, particularly how a steep drop in population size affects the accumulation and purging of deleterious mutations. The Pérez-Pereira model of fitness and dominance coefficients was also utilized to simulate the effects of various conservation scenarios on the population’s viability.
Results: The first simulation scenario examined the impact of a steep population decline on genetic load. The results indicated that a sharp loss in population leads to an increase in genetic load. However, this increase was not very steep due to the rapid purging of recessive deleterious mutations, which caused a significant drop in the proportion of genetic load. Despite this, there was only a moderate increase in the net realized genetic load for recessive deleterious mutations, while dominant deleterious mutations were not removed through purging.
Missed Opportunities:
- The simulation outcomes of ‘survival’ or ‘extinction’ could not be interpreted due to missing labels on the bar plot depicting populations from northern, southern, or combined regions. The stimulation outcome points on the plot, represented in grey and black colors, were not visible, making it difficult to draw conclusions from this data.
- Supplementary figures revealed that over 20 simulated generations with 12 and 24 founders, there was no statistically significant lowering of genetic load, although a trend of lowered genetic load was observed. This suggests that more generations of breeding may be required to achieve a significant reduction in genetic load. Additionally, innovative breeding techniques such as genetic engineering to correct deleterious mutations pre-implantation and in vitro fertilization may be necessary to improve the viability of the Saola population.
- Combining populations for breeding can result in increased genetic diversity and adaptive potential, as well as a decrease in genetic load. However, this approach can also lead to the loss of local adaptability and the introduction of new deleterious mutations into the gene pool. These potential adverse effects, known as outbreeding depression and migration load, were not taken into account in the model.
Conclusion: This study underscores the importance of understanding genetic load within critically endangered species like the Saola. It highlights the need for innovative conservation techniques to address declining populations and improve their long-term survival. While combining populations for breeding may offer benefits such as increased genetic diversity and adaptive potential, it is crucial to consider potential adverse effects like outbreeding depression and migration load. Given the critical status of the Saola, with only a few individuals remaining, breeding the two populations may be the only chance for survival and is the overriding consideration in favor of conservation breeding of Saola populations.
8. DISCUSSION
8.1 Genetic Diversity, Genetic Load and Adaptation: Key Considerations In Conservation Outbreeding
In the realm of plant and animal breeding, particularly within conservation efforts aimed at preserving biodiversity, three fundamental aspects demand careful consideration: genetic diversity, genetic load, and adaptation.
- Genetic Diversity: The variety of genes within a population enables evolution and adaptation. Genetic diversity encompasses the myriad genes present within a population or species, serving as the raw material for evolution and adaptation.
- Genetic Load: Harmful mutations that negatively impact fitness accumulate over generations. Genetic load refers to the accumulation of deleterious mutations that can negatively impact an organism’s fitness, robustness, and reproductive success.
- Adaptation: Introducing beneficial genetic diversity can enhance adaptability and evolutionary potential by providing a broader gene pool from which advantageous traits may emerge or be recombined. However, this process must be navigated judiciously to avoid increasing the detrimental effects of genetic load. Risks include:
- Outbreeding Depression: Introducing too many novel alleles may disrupt locally adapted gene complexes, reducing offspring fitness.
- Migration Load: New alleles might carry hidden deleterious mutations that become expressed under changed conditions, potentially harming population viability.
8.1.1 Balancing Genetic Diversity, Genetic Load and Adaptation
Maintaining a balance between genetic diversity and genetic load is critical for managing breeding populations, as it directly influences the health, adaptability, and long-term survival of species. This equilibrium can be achieved through strategic management that considers environmental stability, selective pressures, and adaptive traits. Genetic diversity exerts both positive and negative influences on a population’s fitness:
- Positive Effects: Increased genetic diversity enhances adaptability to changing environmental conditions by providing a broader range of alleles for natural selection. Higher heterozygosity improves resistance to diseases and reduces the risk of inbreeding depression.
- Negative Effects: Excessive genetic diversity may lead to reduced fitness due to outbreeding depression or increased competition among individuals with high allelic variation. Elevated mutation rates can also result in higher genetic loads, negatively impacting population viability.
To minimize risks and maximize benefits, conservationists must tailor outbreeding strategies to the specific context of each population, carefully balancing the introduction of genetic diversity while preserving existing adaptations.
8.1.2 Enhancing Adaptability and Resilience Due to Genetic Rescue
Adaptability and resilience are two critical advantages conferred by greater genetic diversity within a population. Adaptability is enhanced because a diverse gene pool increases the probability that some individuals will possess traits beneficial for surviving and thriving under new environmental conditions. This genetic variation can be crucial when facing challenges such as emerging diseases, shifting climate patterns, or changes in food availability. Essentially, a genetically diverse population has a broader range of tools at its disposal to cope with and adapt to environmental changes. Resilience, on the other hand, refers to the ability of a population to withstand and recover from environmental stresses and diseases. Higher genetic diversity typically makes populations more robust and less vulnerable to various threats. This phenomenon is often described as the “genetic rescue” effect, where the introduction of new genetic material can revitalize a population suffering from inbreeding depression or other genetic issues. Inbreeding depression occurs when closely related individuals mate, leading to a reduction in genetic diversity and an increase in harmful genetic traits. By introducing new genetic material, the population can overcome these issues, leading to improved health and survival rates. Thus, genetic diversity not only helps populations adapt to new challenges but also enhances their overall resilience and long-term viability.
8.1.3 Loss of Adaptation Due to Outbreeding
Interbreeding between local and external populations can lead to the loss or dilution of adaptive traits crucial for survival in native environments. This phenomenon, known as outbreeding depression, occurs when introduced genetic material disrupts locally adapted gene combinations, reducing overall fitness. For example, uncontrolled crossbreeding in dog breeds has diluted locally adapted traits and reduced genetic diversity within subpopulations. Effective conservation efforts require comprehensive strategies to monitor fitness performance, track allele frequencies, and implement targeted breeding programs. By doing so, conservationists can mitigate outbreeding depression risks while enhancing genetic diversity.
8.2 Simple metricsand strategic consideration for conservation outbreeding
Demography is the study of populations and their characteristics, such as size, growth rate, distribution, and genetic structure. By analyzing these factors, scientists can gain a better understanding of how populations change over time and how they are influenced by various environmental and biological factors.

8.2.1. Effective Population Size (Ne)
Effective population size reflects the number of individuals contributing to the next generation’s gene pool. Importance for Conservation: Effective population size is a measure of how much genetic diversity is maintained in a population. Smaller Ne values indicate greater genetic drift, which can lead to loss of genetic diversity and inbreeding depression. Strategic Outbreeding: When Ne is low, conservation strategies might include introducing genetically diverse individuals from other populations to increase Ne and counteract the effects of genetic drift and inbreeding.
8.2.2. FST Genetic Distance
FST measures genetic differentiation between populations. Importance for Conservation: Effective population size is a measure of how much genetic diversity is maintained in a population. Smaller Ne values indicate greater genetic drift, which can lead to loss of genetic diversity and inbreeding depression. Strategic Outbreeding: When Ne is low, conservation strategies might include introducing genetically diverse individuals from other populations to increase Ne and counteract the effects of genetic drift and inbreeding.
8.2.3. Inbreeding Coefficient (F)
The inbreeding coefficient quantifies the probability of alleles being identical by descent. Importance for Conservation: An elevated inbreeding coefficient often indicates that the breeding pool is too small, leading to the potential for negative genetic effects such as decreased fitness, reduced fertility, and increased genetic disorders. Strategic Outbreeding: By identifying populations with high inbreeding coefficients, conservationists can target these populations for outbreeding to introduce genetic diversity and reduce inbreeding depression.
8.3Complex metricsand strategic consideration for conservation outbreeding
By addressing these interconnected factors, conservationists can develop effective strategies to preserve biodiversity, maintain genetic health, and ensure the long-term survival of vulnerable populations.
8.3.1 Genetic Distance
Introducing genetically distant individuals into a population can significantly increase genetic diversity, which is often beneficial for the overall health and adaptability of the population. This increase in diversity can lead to a more robust population that is better equipped to handle various environmental challenges. However, it may also introduce harmful alleles that could negatively impact the population’s well-being, potentially leading to health issues or reduced fitness. On the other hand, breeding with closely related populations can minimize the risk of introducing harmful mutations, but it may lead to inbreeding depression over time, which can decrease genetic diversity and increase the likelihood of genetic disorders. Conversely, extreme outbreeding introduces more genetic variation, but it also risks disrupting traits that are locally adapted to the environment, which could be detrimental to the population’s survival in its specific habitat.
8.3.2 Selective Pressure
Selective pressures are incredibly important as they play a crucial role in shaping population genetics by influencing which traits are favored in a given environment. Controlled outbreeding can be particularly beneficial for small, inbred populations as it helps to reduce inbreeding depression and increase genetic diversity. This, in turn, enhances the population’s overall resilience, allowing it to better withstand environmental changes and challenges. However, excessive mixing in populations that are already well-adapted to their local environment may disrupt these local adaptations. This disruption can potentially lead to reduced fitness and adaptability, making it difficult for the population to thrive in its natural habitat. The traits that were once advantageous may no longer be present, which can hinder the population’s ability to survive and reproduce successfully.
8.3.3 Mutation Screening
Regular genetic testing is absolutely essential for identifying harmful mutations within a population. This critical process enables conservationists to take proactive and informed measures to mitigate potential risks and maintain the overall genetic health of the population. By diligently screening for mutations, conservationists can develop well-thought-out strategies to manage and preserve the genetic integrity of the population, ensuring that harmful alleles are not passed on to future generations. This proactive and strategic approach helps maintain a healthy gene pool and supports the long-term viability and sustainability of the population.
8.3.4 Fitness Monitoring
Monitoring key indicators such as survival rates, reproductive success, and disease resistance is absolutely crucial. This ensures that breeding practices are effectively maintaining or even improving the overall fitness of the population. If there is any noticeable decline in fitness, it may become necessary to make adjustments to the outbreeding strategies currently being employed to ensure that the population remains healthy and viable. By closely observing these important indicators, conservationists can identify potential issues early on and take corrective action to support the population’s continued success and well-being.
8.3.5 Environmental Stability
Stable environments generally tend to favor the development and maintenance of local adaptations. These adaptations are crucial because they allow populations to thrive and succeed in their specific habitats, ensuring that they are well-suited to the particular conditions they encounter. However, when conditions are changing, it may become necessary to introduce external genes to enhance adaptability. Conservationists must carefully evaluate the stability of the environment to determine whether it is more important to prioritize preserving existing adaptations or to introduce new genetic variation to cope with changing conditions. This careful evaluation is essential, as it helps ensure that the population can continue to survive and adapt in the face of environmental changes, maintaining a balance between stability and adaptability.
8.3.6 Breeding Strategies and Genetic Drift
Balancing genetic variation is a crucial aspect of conservation efforts, which involves the careful selection of mating pairs based on their relatedness and allelic diversity. The primary goal is to avoid the pitfalls of excessive inbreeding or outbreeding, which can have detrimental effects on a population’s genetic health. Conservationists have a range of techniques at their disposal, including captive breeding programs, genetic engineering methods, and assisted migration strategies, all aimed at enhancing genetic diversity within a population. These approaches are essential in minimizing the risks associated with genetic drift and non-random mating patterns, which can threaten the long-term survival and adaptability of the population. By meticulously managing breeding strategies, conservationists can effectively support the population’s genetic health and resilience, thereby promoting its ability to thrive and adapt to changing environments in the future.
8.4 Practical Rules for Conservation Strategic Outbreeding
Determining safe thresholds for factors influencing outbreeding dynamics is essential when managing breeding programs for endangered species with limited genomic resources (e.g., draft genomes). For instance, when working with two endangered species with only draft genome sequences, evaluating critical aspects of outbreeding is necessary. To effectively manage genetic diversity and genetic load in small, isolated populations, the following practical rules should be followed:
8.4.1 Encourage Controlled Outbreeding: For small or inbred populations showing signs of inbreeding depression (reduced fitness due to recessive deleterious mutations) or genetic disorders, controlled outbreeding can increase genetic diversity and reduce the negative effects of excessive homozygosity. Introducing novel alleles from external sources can enhance population resilience and confer adaptive advantages. However, this approach must be carefully managed to avoid introducing harmful mutations that could negatively impact fitness.
8.4.2 Limit Outbreeding in Specific Cases: When dealing with highly adapted local populations or external populations carrying harmful traits, outbreeding should be limited. This precaution prevents the introduction of deleterious mutations and preserves the fitness of the native population. Outbreeding depression, or migration load, can occur when foreign genetic material disrupts locally adaptive gene combinations, reducing fitness in the native environment.
8.4.3 Risk Assessment and Strategic Management: Maintaining a balance between genetic diversity and genetic load requires continuous monitoring and management. While increased diversity can introduce beneficial alleles, it may also elevate genetic load, potentially harming fitness. Conservationists must carefully assess relationships, pressures, mutations, and environmental stability to design strategic breeding programs tailored to each population’s context. Using draft genomes, focusing on metrics like FST, F, Ne, and mutation screening, and closely monitoring fitness can help make informed decisions. Adjusting thresholds based on empirical data ensures a balanced approach that maximizes genetic diversity while minimizing risks, promoting biodiversity preservation and long-term survival.
8.5 Conservation: Outbreeding Decisions and Results Example
A practical example involves the Florida panther (Puma concolor coryi), which faced severe genetic bottlenecks leading to low Ne (~20). To mitigate this, conservationists introduced eight female Texas cougars (Puma concolor stanleyana) into the Florida panther population to increase genetic diversity and reduce inbreeding coefficients (initially around 0.25). Post-introduction studies showed an increase in Ne and a decrease in F, demonstrating successful outbreeding as a conservation strategy.
Another example is the red wolf (Canis rufus), which experienced a severe decline leading to a genetic bottleneck. Conservation efforts included outbreeding with coyotes (Canis latrans) to increase genetic diversity. However, this has been a complex issue, as hybridization with coyotes can threaten the genetic integrity of the red wolf. Careful management and monitoring are crucial in this case to ensure that the introduced individuals do not disrupt local adaptations or introduce maladaptive genes.
Similarly, Przewalski’s horse (Equus ferus przewalskii), once extinct in the wild, was reintroduced through captive breeding programs. To enhance genetic diversity and improve the long-term viability of the reintroduced population, individuals from different captive lineages were crossbred through outbreeding strategies (International Union for Conservation of Nature [IUCN], 2024). Despite ths success, challenges such as nbreeding, hybridization with domestic horses and genetic management remain ongoing concern for conservation.
These successes demonstrate that conservation outbreeding can be an effective tool for managing endangered species populations and improving their long-term viability. However, careful management and monitoring are necessary to ensure that the introduced individuals do not disrupt local adaptations or introduce maladaptive genes.
8.6Proposed Workflow for In Vitro Innovation for Quick Multi-Generational Expansion of Vulnerable and Endangered Species
8.6.1 Field Work: Sample Collection for Scientific Research
Humans face a complex ethical and practical dilemma regarding wildlife conservation. While the ideal scenario involves minimal human interference in natural ecosystems, the reality of human-caused threats, such as poaching and deforestation, necessitates intervention to prevent species extinction. This intervention requires a careful balance to minimize harm while maximizing conservation efforts. Any intervention strategy must prioritize ethical considerations and adhere to best practices. This includes minimizing stress and harm to animals, respecting their natural behaviors, and ensuring that interventions are scientifically sound and evidence-based. The use of tranquilization and sample collection for scientific research should be conducted with the utmost care and expertise.
The involvement of experienced wildlife field scientists and conservationists is crucial for responsible intervention. Individuals like Steve Backshall, known for his work on the Deadly 60 TV show, exemplify the type of expertise needed. These professionals possess a deep understanding of animal behavior, ecology, and conservation principles. They are trained in humane handling techniques, minimizing stress and risk to the animals. Their work often involves collaboration with other scientists, conservation organizations, and government agencies to ensure that interventions are effective and aligned with conservation goals. The legacy of figures like Steve Irwin, Dame Jane Goodall, and Sir David Attenborough underscores the importance of combining scientific knowledge with a profound respect for nature. Their approach emphasizes observation, understanding, and a commitment to conservation, rather than exploitation or unnecessary disturbance.
The collection of wildlife samples for scientific research is a critical tool for understanding species and developing effective conservation strategies. Genetic analysis, disease monitoring, and physiological studies can provide valuable insights into population health, threats, and potential interventions. However, this process must be conducted ethically and responsibly. Permits and approvals from relevant authorities are essential, and all procedures should adhere to strict animal welfare guidelines. The use of minimally invasive techniques, such as non-lethal sampling methods, is preferred whenever possible.
8.6.2 Lab Work: Preparation of Stem Cells from Collected Samples for in vitro conservation breeding
One of the hurdles of conservation is limitation of individuals available for breeding and generation length i.e. long time is taken for gestation and newborn animal to mature, and give birth to next generation. The objective of this workflow is to reduce generation time and increase the yield of individuals produced per generation in Saola models. This protocol outlines a putative laboratory procedure that begins with various reproductive materials, including sperm, eggs, blastocysts, embryonic stem cells (ESCs), or induced pluripotent stem cells (iPSCs). The process involves multiple cycles of fertilization and germ cell development, culminating in the implantation of a final blastocyst, which is subsequently allowed to mature into an adult Saola.
- Selection of Starting Materials: The workflow commences with the selection of starting materials. Sperm can be harvested from the epididymis or vas deferens of a donor Saola, while eggs are obtained from superovulated female Saola. Additionally, blastocysts may be collected from early-stage embryos, and ESCs or iPSCs can be derived from either blastocysts or reprogrammed somatic cells. IPSCs are obtained from somatic cells such as skin cells, blood cells, hair follicle, fat tissue, bone marrow, sperms and eggs because these regions have natural regenerative potential in adult mammals due to presence of adult stem cells that provide repair and regeneration function naturally in the sites they occur by differentiating into mature tissue cells.
- Fertilization Techniques: Following the collection of these materials, the next step involves fertilization, which can be achieved through in vitro fertilization (IVF) or intracytoplasmic sperm injection (ICSI). These techniques facilitate the creation of zygotes, which are fertilized eggs that will develop into blastocysts in vitro.
- Embryonic Stem Cell Culture: Subsequently, embryonic stem cells are extracted from the inner cell mass of the blastocysts and cultured under specific conditions that preserve their pluripotency, allowing them to differentiate into various cell types. The next phase involves differentiating these ESCs into primordial germ cells (PGCs) using established protocols, which are then matured into functional sperm and eggs within specialized culture systems.
- Repetition of the Cycle: With the lab-grown sperm and eggs, another round of IVF or ICSI is performed, leading to the generation of new zygotes that will again develop into blastocysts. This cycle of deriving ESCs, differentiating them into germ cells, and conducting fertilization is repeated for the desired number of generations, typically around five. Each cycle can be completed in approximately two to three weeks, contingent upon the efficiency of the protocols employed.
- Final Implantation and Development: Upon reaching the final generation, the last blastocyst is implanted into a surrogate female Saola utilizing a nonsurgical embryo transfer technique. The embryo is then allowed to develop to term, ultimately resulting in the maturation of an adult Saola.
Throughout this workflow, key considerations include monitoring for epigenetic stability, assessing the health and viability of embryos and germ cells at each developmental stage, and ensuring that all procedures are conducted in a specified pathogen-free (SPF) environment to promote reproducibility and uphold animal welfare standards.
9. REFERENCES
9.1 References for Introduction section
Deasly 60 Rhino Rescue Special | Host Steve Backshall https://www.imdb.com/title/tt19719186/
How Deforestation Looks From Space | Earth From Space | BBC Earth https://youtu.be/ctlUvcKhsSg
Chung, Mi Yoon, Juha Merilä, Jialiang Li, Kangshan Mao, Jordi López-Pujol, Yoshihiko Tsumura, and Myong Gi Chung. 2023. “Neutral and Adaptive Genetic Diversity in Plants: An Overview.” Frontiers in Ecology and Evolution 11 (February). https://doi.org/10.3389/fevo.2023.1116814.
“Genetic Load – ScienceDirect.” n.d. Accessed June 7, 2025. https://www.sciencedirect.com/science/article/abs/pii/S0960982224015112.
“Last Chance to Save Saola from Extinction – IUCN | IUCN.” 2009. September 3, 2009. https://iucn.org/content/last-chance-save-saola-extinction-iucn.
Lou, Jiao, Weina Li, Panlong Chen, Haiyan Chen, Amna Shakoor, Yunlong Chen, Jinlian Hua, Yan Wang, and Shiqiang Zhang. 2025. “Application of Induced Pluripotent Stem Cells in the Conservation of Endangered Animals.” Stem Cell Research & Therapy 16 (1): 261. https://doi.org/10.1186/s13287-025-04392-5.
Pearman, Peter B., Olivier Broennimann, Tsipe Aavik, Tamer Albayrak, Paulo C. Alves, F. A. Aravanopoulos, Laura D. Bertola, et al. 2024. “Monitoring of Species’ Genetic Diversity in Europe Varies Greatly and Overlooks Potential Climate Change Impacts.” Nature Ecology & Evolution 8 (2): 267–81. https://doi.org/10.1038/s41559-023-02260-0.
“Pseudoryx Nghetinhensis (Saola).” n.d. Animal Diversity Web. Accessed June 7, 2025. https://animaldiversity.org/accounts/Pseudoryx_nghetinhensis/.
Salgotra, Romesh Kumar, and Bhagirath Singh Chauhan. 2023. “Genetic Diversity, Conservation, and Utilization of Plant Genetic Resources.” Genes 14 (1): 174. https://doi.org/10.3390/genes14010174.
“Saola | Species | WWF.” n.d. World Wildlife Fund. Accessed June 7, 2025. https://www.worldwildlife.org/species/saola.
Seale, Nina. 2023. “One Last Chance to Find the Saola.” Synchronicity Earth (blog). October 25, 2023. https://www.synchronicityearth.org/one-last-chance-saola/.
“Starting Materials For iPSCs.” n.d. Accessed June 7, 2025. https://www.cellandgene.com/doc/starting-materials-for-ipscs-0001.
“Wild ActionFormer: Enhancing Wildlife Action Recognition for 11 Endangered Species in Wolong.” 2025. Ecological Informatics 89 (November):103148. https://doi.org/10.1016/j.ecoinf.2025.103148.
9.2 References for Methods section
Camargo, Arley. 2022. “PCAtest: Testing the Statistical Significance of Principal Component Analysis in R.” PeerJ 10 (February):e12967. https://doi.org/10.7717/peerj.12967.
Ceballos, Francisco C., Peter Joshi, David Clark, Michele Ramsay, and James Wilson. 2018. “Runs of Homozygosity: Windows into Population History and Trait Architecture.” Nature Reviews Genetics, January. https://doi.org/10.1038/nrg.2017.109.
Chen, Wanzhao, Ronghe Xing, Panpan Xia, Yujie Yang, Chao Ma, and Lining Xia. 2025. “Whole Genome Sequencing Informs SNP-Based Breeding Strategies to Safeguard Genetic Diversity in Captive African Lions.” Frontiers in Veterinary Science 12 (April). https://doi.org/10.3389/fvets.2025.1577726.
Drillon, Guénola, Raphaël Champeimont, Francesco Oteri, Gilles Fischer, and Alessandra Carbone. 2020. “Phylogenetic Reconstruction Based on Synteny Block and Gene Adjacencies.” Molecular Biology and Evolution 37 (9): 2747–62. https://doi.org/10.1093/molbev/msaa114.
Dutta, Abhishek, Fabien Dutreux, and Joseph Schacherer. 2021. “Loss of Heterozygosity Results in Rapid but Variable Genome Homogenization across Yeast Genetic Backgrounds.” Edited by Antonis Rokas and Huda Y Zoghbi. eLife 10 (June):e70339. https://doi.org/10.7554/eLife.70339.
Forkman, Johannes, Julie Josse, and Hans-Peter Piepho. 2019. “Hypothesis Tests for Principal Component Analysis When Variables Are Standardized.” Journal of Agricultural, Biological and Environmental Statistics 24 (2): 289–308. https://doi.org/10.1007/s13253-019-00355-5.
“Genome-Wide Detection of Runs of Homozygosity and Heterozygosity in Tunchang Pigs.” 2024. Animal 18 (8): 101236. https://doi.org/10.1016/j.animal.2024.101236.
Gislason, Hannes. 2023. “SNP Heterozygosity, Relatedness and Inbreeding of Whole Genomes from the Isolated Population of the Faroe Islands.” BMC Genomics 24 (1): 707. https://doi.org/10.1186/s12864-023-09763-x.
Graf, Louis, Younhee Shin, Ji Hyun Yang, Ji Won Choi, Il Ki Hwang, Wendy Nelson, Debashish Bhattacharya, Frédérique Viard, and Hwan Su Yoon. 2021. “A Genome-Wide Investigation of the Effect of Farming and Human-Mediated Introduction on the Ubiquitous Seaweed Undaria Pinnatifida.” Nature Ecology & Evolution 5 (3): 360–68. https://doi.org/10.1038/s41559-020-01378-9.
Guo, Yan, Fei Ye, Quanghu Sheng, Travis Clark, and David C. Samuels. 2014. “Three-Stage Quality Control Strategies for DNA Re-Sequencing Data.” Briefings in Bioinformatics 15 (6): 879–89. https://doi.org/10.1093/bib/bbt069.
Ha, Young-Ho, Jong-Bin An, Jaesang Chung, Jung-Won Yoon, and Hee-Young Gil. 2025. “Genotyping-by-Sequencing Reveals Low Genetic Diversity and Peripheral Isolation in Southern Populations of Sophora Koreensis, a Korean Endemic Shrub.” Scientific Reports 15 (1): 17996. https://doi.org/10.1038/s41598-025-02703-7.
Hall, David, Wei Zhao, Ulfstand Wennström, Bengt Andersson Gull, and Xiao-Ru Wang. 2020. “Parentage and Relatedness Reconstruction in Pinus Sylvestris Using Genotyping-by-Sequencing.” Heredity 124 (5): 633–46. https://doi.org/10.1038/s41437-020-0302-3.
Jiang, Wei, Xiangyu Zhang, Siting Li, Shuang Song, and Hongyu Zhao. 2022. “An Unbiased Kinship Estimation Method for Genetic Data Analysis.” BMC Bioinformatics 23 (1): 525. https://doi.org/10.1186/s12859-022-05082-2.
Jiang, Yao, Xiaojin Li, Jiali Liu, Wei Zhang, Mei Zhou, Jieru Wang, Linqing Liu, et al. 2022. “Genome-Wide Detection of Genetic Structure and Runs of Homozygosity Analysis in Anhui Indigenous and Western Commercial Pig Breeds Using PorcineSNP80k Data.” BMC Genomics 23 (1): 373. https://doi.org/10.1186/s12864-022-08583-9.
Kim, Su Yeon, Kirk E. Lohmueller, Anders Albrechtsen, Yingrui Li, Thorfinn Korneliussen, Geng Tian, Niels Grarup, et al. 2011. “Estimation of Allele Frequency and Association Mapping Using Next-Generation Sequencing Data.” BMC Bioinformatics 12 (1): 231. https://doi.org/10.1186/1471-2105-12-231.
Lampi, Sara, Jonas Donner, Heidi Anderson, and Jaakko Pohjoismäki. 2020. “Variation in Breeding Practices and Geographic Isolation Drive Subpopulation Differentiation, Contributing to the Loss of Genetic Diversity within Dog Breed Lineages.” Canine Medicine and Genetics 7 (1): 5. https://doi.org/10.1186/s40575-020-00085-9.
Laurie, Cathy C., Kimberly F. Doheny, Daniel B. Mirel, Elizabeth W. Pugh, Laura J. Bierut, Tushar Bhangale, Frederick Boehm, et al. 2010. “Quality Control and Quality Assurance in Genotypic Data for Genome-Wide Association Studies.” Genetic Epidemiology 34 (6): 591–602. https://doi.org/10.1002/gepi.20516.
Li, Rong, Shanyuan Chen, Chunqing Li, Heng Xiao, Vânia Costa, Mohammad Shamsul Alam Bhuiyan, Mumtaz Baig, and Albano Beja-Pereira. 2022. “Whole-Genome Analysis Deciphers Population Structure and Genetic Introgression Among Bovine Species.” Frontiers in Genetics 13 (May). https://doi.org/10.3389/fgene.2022.847492.
Liu, Shanlin, Michael V. Westbury, Nicolas Dussex, Kieren J. Mitchell, Mikkel-Holger S. Sinding, Peter D. Heintzman, David A. Duchêne, et al. 2021. “Ancient and Modern Genomes Unravel the Evolutionary History of the Rhinoceros Family.” Cell 184 (19): 4874-4885.e16. https://doi.org/10.1016/j.cell.2021.07.032.
Ma, Yiqing, Fangrui Lou, Xiaofei Yin, Bailin Cong, Shenghao Liu, Linlin Zhao, and Li Zheng. 2022. “Whole-Genome Survey and Phylogenetic Analysis of Gadus Macrocephalus.” Bioscience Reports 42 (7): BSR20221037. https://doi.org/10.1042/BSR20221037.
MacHugh, David E, Mark D Shriver, Ronan T Loftus, and Patrick Cunningham. n.d. “hDomestication and Phylogeography of Taurine and Zebu Cattle (BOSt u u m and Bos Indicus).”
Meyermans, R., W. Gorssen, N. Buys, and S. Janssens. 2020. “How to Study Runs of Homozygosity Using PLINK? A Guide for Analyzing Medium Density SNP Data in Livestock and Pet Species.” BMC Genomics 21 (1): 94. https://doi.org/10.1186/s12864-020-6463-x.
Minn, Aye Ko Ko, Motomichi Matsuzaki, Akira Narita, Takamitsu Funayama, Yurii Kotsar, Satoshi Makino, Jun Takayama, Shinichi Kuriyama, and Gen Tamiya. 2025. “Profiling of Runs of Homozygosity from Whole-Genome Sequence Data in Japanese Biobank.” Journal of Human Genetics 70 (6): 287–96. https://doi.org/10.1038/s10038-025-01331-3.
Mualim, Kristy, Christoph Theunert, and Montgomery Slatkin. 2021. “Estimation of Coalescence Probabilities and Population Divergence Times from SNP Data.” Heredity 127 (1): 1–9. https://doi.org/10.1038/s41437-021-00435-8.
Nelson, Holly V., Luke Silver, Toby G. L. Kovacs, Elspeth A. McLennan, Arthur Georges, Jane L. DeGabriel, Carolyn J. Hogg, and Katherine Belov. 2025. “Genome-Wide Diversity and MHC Characterisation in a Critically Endangered Freshwater Turtle Susceptible to Disease.” Immunogenetics 77 (1): 21. https://doi.org/10.1007/s00251-025-01378-8.
Pavan, Stefano, Chiara Delvento, Luigi Ricciardi, Concetta Lotti, Elena Ciani, and Nunzio D’Agostino. 2020. “Recommendations for Choosing the Genotyping Method and Best Practices for Quality Control in Crop Genome-Wide Association Studies.” Frontiers in Genetics 11 (June). https://doi.org/10.3389/fgene.2020.00447.
Purcell, Shaun, Benjamin Neale, Kathe Todd-Brown, Lori Thomas, Manuel A. R. Ferreira, David Bender, Julian Maller, et al. 2007. “PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses.” American Journal of Human Genetics 81 (3): 559–75. https://doi.org/10.1086/519795.
Purfield, Deirdre C, Donagh P Berry, Sinead McParland, and Daniel G Bradley. 2012. “Runs of Homozygosity and Population History in Cattle.” BMC Genetics 13 (August):70. https://doi.org/10.1186/1471-2156-13-70.
“Quantitative Trait Mapping: Calculating A Kinship Matrix.” n.d. Accessed June 7, 2025. https://smcclatchy.github.io/mapping/08-calc-kinship/.
“Rate of Coalescence of Lineage Pairs in the Spatial Λ-Fleming–Viot Process.” 2022. Theoretical Population Biology 146 (August):15–28. https://doi.org/10.1016/j.tpb.2022.05.002.
Schmidt, Thomas L., Moshe-Elijah Jasper, Andrew R Weeks, and Ary A Hoffmann. 2021. “Unbiased Population Heterozygosity Estimates from Genome-Wide Sequence Data.” Methods in Ecology and Evolution 12 (10): 1888–98. https://doi.org/10.1111/2041-210X.13659.
Silva, Gabriel A. A., Avril M. Harder, Kenneth B. Kirksey, Samarth Mathur, and Janna R. Willoughby. 2024. “Detectability of Runs of Homozygosity Is Influenced by Analysis Parameters and Population-Specific Demographic History.” PLOS Computational Biology 20 (10): e1012566. https://doi.org/10.1371/journal.pcbi.1012566.
Soltis, Pamela S., and Douglas E. Soltis. 2003. “Applying the Bootstrap in Phylogeny Reconstruction.” Statistical Science 18 (2). https://doi.org/10.1214/ss/1063994980.
Tavaré, Simon, David J. Balding, R. C. Griffiths, and Peter Donnelly. 1997. “Inferring Coalescence Times From DNA Sequence Data.” Genetics 145 (2): 505–18. https://doi.org/10.1093/genetics/145.2.505.
Upadhyay, M R, W Chen, J A Lenstra, C R J Goderie, D E MacHugh, S D E Park, D A Magee, et al. 2017. “Genetic Origin, Admixture and Population History of Aurochs (Bos Primigenius) and Primitive European Cattle.” Heredity 118 (2): 169–76. https://doi.org/10.1038/hdy.2016.79.
Xu, Zhong, Shuqi Mei, Jiawei Zhou, Yu Zhang, Mu Qiao, Hua Sun, Zipeng Li, et al. 2021. “Genome-Wide Assessment of Runs of Homozygosity and Estimates of Genomic Inbreeding in a Chinese Composite Pig Breed.” Frontiers in Genetics 12 (September). https://doi.org/10.3389/fgene.2021.720081.
Yang, Lin, Hong Jin, Qien Yang, Andrey Poyarkov, Miroslav Korablev, Viatcheslav Rozhnov, Junjie Shao, et al. 2025. “Genomic Evidence for Low Genetic Diversity but Purging of Strong Deleterious Variants in Snow Leopards.” Genome Biology 26 (1): 94. https://doi.org/10.1186/s13059-025-03555-0.
9.3 References for Discussion section
Damnjanović, Dragomir, Masoud Nazarizadeh, Monika M. Wisniewska, Václav Pavel, Bohumír Chutný, Arild Johnsen, Milena Nováková, and Jan Štefka. 2024. “Limited Genetic Depletion despite Extinction Risk: Genomic Diversity of a Peripheral Population of Red-Spotted Bluethroats in Central Europe.” Zoological Journal of the Linnean Society 201 (4). https://doi.org/10.1093/zoolinnean/zlae094.
“Effects of Migration Load, Selfing, Inbreeding Depression, and the Genetics of Adaptation on Autotetraploid versus Diploid Establishment in Peripheral Habitats | Evolution | Oxford Academic.” n.d. Accessed June 7, 2025. https://academic.oup.com/evolut/article/75/1/39/6730375.
Flack, Nicole, Lauren Hughes, Jacob Cassens, Maya Enriquez, Samrawit Gebeyehu, Mohammed Alshagawi, Jason Hatfield, et al. 2024. “The Genome of Przewalski’s Horse (Equus Ferus Przewalskii).” G3 Genes|Genomes|Genetics 14 (8). https://doi.org/10.1093/g3journal/jkae113.
Frankham, Richard. 1999. “Quantitative Genetics in Conservation Biology.” Genetics Research 74 (3): 237–44. https://doi.org/10.1017/S001667239900405X.
Frankham, Richard. 2005. “Genetics and Extinction.” Biological Conservation 126 (2): 131–40. https://doi.org/10.1016/j.biocon.2005.05.002.
Gese, Eric M., Fred F. Knowlton, Jennifer R. Adams, Karen Beck, Todd K. Fuller, Dennis L. Murray, Todd D. Steury, Michael K. Stoskopf, Will T. Waddell, and Lisette P. Waits. 2015. “Managing Hybridization of a Recovering Endangered Species: The Red Wolf Canis Rufus as a Case Study.” Current Zoology 61 (1): 191–205. https://doi.org/10.1093/czoolo/61.1.191.
Gong, Guang-Nan, Yuan Wang, Zhi-Ying Zhu, Yi Wang, Elvira Hörandl, Xiao-Ru Wang, Zhi-Qing Xue, and Li He. 2025. “Evolutionary Population Dynamics and Conservation Strategies for Salix Baileyi – a Species with Extremely Small Populations.” Global Ecology and Conservation 58 (April):e03504. https://doi.org/10.1016/j.gecco.2025.e03504.
Griswold, Cortland K. 2021. “The Effects of Migration Load, Selfing, Inbreeding Depression, and the Genetics of Adaptation on Autotetraploid versus Diploid Establishment in Peripheral Habitats.” Evolution 75 (1): 39–55. https://doi.org/10.1111/evo.14127.
Hull, Kelvin L., Matthew P. Greenwood, Melissa Lloyd, Marissa Brink-Hull, Aletta E. Bester-van der Merwe, and Clint Rhode. n.d. “Drivers of Genomic Diversity and Phenotypic Development in Early Phases of Domestication in Hermetia Illucens.” Accessed June 7, 2025. https://doi.org/10.1111/imb.12940.
Johnson, Warren E., David P. Onorato, Melody E. Roelke, E. Darrell Land, Mark Cunningham, Robert C. Belden, Roy McBride, et al. 2010. “Genetic Restoration of the Florida Panther.” Science, September. https://doi.org/10.1126/science.1192891.
Kuang, Weimin, Jingyang Hu, Hong Wu, Xiaotian Fen, Qingyan Dai, Qiaomei Fu, Wen Xiao, et al. 2020. “Genetic Diversity, Inbreeding Level, and Genetic Load in Endangered Snub-Nosed Monkeys (Rhinopithecus).” Frontiers in Genetics 11 (December). https://doi.org/10.3389/fgene.2020.615926.
Lovrenčić, Leona, Martina Temunović, Lena Bonassin, Frederic Grandjean, Christopher M. Austin, and Ivana Maguire. 2022. “Climate Change Threatens Unique Genetic Diversity within the Balkan Biodiversity Hotspot – The Case of the Endangered Stone Crayfish.” Global Ecology and Conservation 39 (November):e02301. https://doi.org/10.1016/j.gecco.2022.e02301.
Mathur, Samarth, John M. Tomeček, Luis A. Tarango-Arámbula, Robert M. Perez, and J. Andrew DeWoody. 2023. “An Evolutionary Perspective on Genetic Load in Small, Isolated Populations as Informed by Whole Genome Resequencing and Forward-Time Simulations.” Evolution 77 (3): 690–704. https://doi.org/10.1093/evolut/qpac061.
Pantoja, Pauline O., C. E. Timothy Paine, and Mario Vallejo-Marín. 2018. “Natural Selection and Outbreeding Depression Suggest Adaptive Differentiation in the Invasive Range of a Clonal Plant.” Proceedings of the Royal Society B: Biological Sciences, July. https://doi.org/10.1098/rspb.2018.1091.
Sachdeva, Himani, Oluwafunmilola Olusanya, and Nick Barton. 2022. “Genetic Load and Extinction in Peripheral Populations: The Roles of Migration, Drift and Demographic Stochasticity.” Philosophical Transactions of the Royal Society B, March. https://doi.org/10.1098/rstb.2021.0010.
Turghan, Mardan Aghabey, Zhigang Jiang, and Zhongze Niu. 2022. “An Update on Status and Conservation of the Przewalski’s Horse (Equus Ferus Przewalskii): Captive Breeding and Reintroduction Projects.” Animals 12 (22): 3158. https://doi.org/10.3390/ani12223158.
10. DEFINITIONS
Genome: The genome represents the comprehensive set of deoxyribonucleic acid (DNA) within an organism, encompassing all its genes and hereditary information. In eukaryotic organisms such as humans and plants, the genome comprises two sets of chromosomes – one inherited from each parent through sexual reproduction. This diploid nature allows for genetic diversity and facilitates Mendelian inheritance patterns. The human reference genome, serving as a standard sequence against which other genomes are compared, contains approximately 3 billion base pairs distributed across three distinct types: adenine (A), thymine (T), cytosine (C), and guanine (G). These four nucleotides form the building blocks of DNA molecules through specific pairing rules – adenine always pairs with thymine via two hydrogen bonds, while cytosine pairs with guanine using three hydrogen bonds. This base pairing enables the replication and transcription processes that maintain genetic integrity across generations. To elucidate this concept through analogy, consider a genome as akin to an extensive cookbook collection containing all our DNA combined, which collectively defines us as individuals or species within the realm of biology. In humans specifically, this ‘collection’ comprises roughly 3 billion recipes (DNA base pairs), each composed of one of four possible bases: adenine (A), thymine (T), cytosine (C), and guanine (G).
Genetic Sequencing: Genetic sequencing encompasses the suite of laboratory techniques and technologies employed to determine the precise order of nucleotides – adenine (A), thymine (T), cytosine (C), guanine (G) – within an organism’s genome. This critical process enables researchers to elucidate genetic information at both population and species levels, fostering a deeper understanding of biological systems. Several advanced sequencing methods exist, including:
1. Next-Generation Sequencing (NGS): A high-throughput approach that allows for rapid and parallel sequencing of millions of DNA fragments simultaneously.
2. Sanger Sequencing: The first method developed for DNA sequencing, involving chain termination reactions to determine the sequence of nucleotides in a given template strand.
3. Shotgun Sequencing: An unbiased strategy where an entire genome is randomly fragmented into smaller pieces before being sequenced individually; this approach facilitates de novo assembly of genomes without prior reference sequences.
The data generated through genetic sequencing serves multiple purposes:
– Genetic Diversity Analysis: By comparing DNA sequences from different individuals or populations, researchers can assess levels of genetic variation and infer evolutionary histories.
– Allele Identification: Sequencing enables the identification of specific alleles associated with traits of interest, such as disease susceptibility genes or adaptive variants under selection.
– Phylogenetic Analyses: Comparative sequencing across species allows for reconstructing evolutionary relationships through phylogenetic tree construction, shedding light on speciation events and taxonomic classifications.
– Genome-Wide Association Studies (GWAS): Sequencing data facilitates large-scale investigations into the molecular basis behind complex phenotypes or diseases by identifying genetic variants associated with these traits across entire genomes.
In summary, genetic sequencing represents a powerful toolbox for unraveling the intricate details of genomic architecture, function, and evolution within and among species.
A gene is the fundamental physical and functional unit of heredity, comprising a specific length of DNA that encodes for either a particular protein or RNA molecule, influencing an organism’s traits (phenotype). Genes are situated on chromosomes, which consist of lengthy strands of deoxyribonucleic acid (DNA) in eukaryotic organisms. To further illustrate this idea using analogy, genes can be likened to recipes that guide our cells in producing proteins – the building blocks for our bodies. They comprise DNA sequences akin to specific steps or ingredients and reside on chromosomes, which are long strings within cell nuclei, much like cookbooks present in each kitchen.
Alleles: Alleles are distinct versions of the same gene occupying a specific location (locus) on homologous DNA molecules within an organism’s genome. They account for diverse phenotypic traits such as eye color and blood type by encoding slightly different proteins or influencing gene expression differentially. In diploid organisms, each individual carries two copies of autosomal genes – one maternal allele and one paternal allele at the same locus on homologous pairs of chromosomes. However, sex-linked genes exhibit a notable exception; males possess only a single copy due to their hemizygous nature. To illustrate this concept via analogy, alleles can be envisioned as different versions of recipes that may yield slightly varied outcomes when followed by cooks – much like substituting apples for bananas could alter the taste of your pie. Typically, we inherit two copies (one from our mother and one from our father) for most traits; however, some genes occur in only a single copy due to their location on sex chromosomes or other factors influencing gene dosage. This allelic variation allows for genetic diversity within populations and enables natural selection to act upon advantageous alleles over generations. Furthermore, the interaction between different alleles at multiple loci can result in complex trait inheritance patterns, as observed in polygenic traits such as height or intelligence.
Genetic Variation: This term denotes the disparities in gene frequencies that exist both within and between populations. Such variability is indispensable for a species’ adaptive capacity, as it furnishes natural selection with raw material upon which to act. The origination of this variation can be attributed to three primary processes: mutations (alterations in DNA sequence), genetic recombination occurring during sexual reproduction, and the migration of individuals among distinct populations. To elucidate this concept through analogy, consider a box filled with differently colored beads. Each color represents an allele, which are variants of genes – the instructions governing our bodily traits such as eye or hair color. These beads originate from two parents and blend during reproduction to generate unique combinations in their offspring, thereby fostering diversity within populations.
Genetic Diversity (long definition): Genetic diversity, alternatively referred to as gene pool variation or population-level genetic variability, denotes the aggregate number of distinct alleles present within a species’ populations. This crucial aspect of biological systems is influenced by several factors, including mutation rates introducing novel alleles and migration patterns facilitating allele exchange between subpopulations within an ecosystem. The significance of genetic diversity lies in its correlation with enhanced adaptive potential for survival under fluctuating environmental conditions or disease pressures. Populations possessing higher levels of genetic variability are better equipped to withstand selective pressures and maintain long-term viability due to their increased capacity for natural selection, genetic drift, and evolutionary adaptation. To illustrate this concept via analogy, envision a group of individuals each holding unique cookbooks – these represent the genetic diversity within a population. The greater the variety in their collective ‘cookbook’ collection, the more adept they become at adapting to changes such as new recipes introduced through migration or shifts in environmental conditions. This increased adaptability arises from having access to diverse alleles that encode for varied traits and physiological responses. In essence, genetic diversity serves as a reservoir of potential adaptations upon which natural selection can act, thereby promoting population resilience and evolutionary success over time.
Genetic Load: Genetic load refers to the decrement in a population’s average fitness resulting from the presence of deleterious alleles within its gene pool, compared to an idealized state comprising solely optimal (non-deleterious) traits. This measure reflects the burden imposed by harmful genetic variants on a population’s overall reproductive success and survival. The concept of genetic load underscores how natural selection operates against detrimental mutations, ultimately leading to their gradual elimination from populations over time through processes such as purifying selection. As these deleterious alleles are removed or reduced in frequency due to selective pressures, they contribute to evolutionary phenomena like adaptation and speciation events by shaping the trajectory of genetic change within a species. To elucidate this concept via analogy, consider genetic load as akin to having some poor-quality ingredients (deleterious alleles) mixed into our cookbooks. While these subpar components may not yield exceptional dishes on their own, they remain present in the gene pool nonetheless. Over time and through repeated trials of various recipes – a metaphor for natural selection’s process of evaluating different genotypes under varying environmental conditions – those less-than-ideal genes can be gradually weeded out or reduced to low frequencies within populations. In essence, genetic load represents an ongoing challenge faced by populations as they strive towards evolutionary optimization, with natural selection serving as the primary mechanism driving the purging of harmful alleles from gene pools.
Genotype Classification: Individuals can exhibit different genetic constitutions, or genotypes, at a particular locus due to the presence of distinct alleles. Two primary categories exist: homozygous and heterozygous.
1. Homozygosity: An individual is considered homozygotic for a specific gene when they possess two identical alleles at that locus on both homologous chromosomes. This can occur in one of two ways:
– AA (homozygous dominant): Both copies are functional, resulting in the expression of the dominant trait.
– aa (homozygous recessive): Neither copy is functional due to mutations or deletions; thus, only the recessive phenotype manifests.
– Using an analogy: Homozygosity resembles having two identical cookbooks. If both versions contain the same recipe for a dish, preparing it will yield consistent results each time – either desirable or undesirable depending on the trait under consideration and whether dominance is involved.
2. Heterozygosity: An individual displays heterozygosity at a specific locus when they carry two different alleles on their homologous chromosomes, denoted as (Aa). The resulting phenotype depends largely upon the dominance relationships between these alleles:
– Incomplete Dominance: Heterozygotes exhibit an intermediate or blended trait compared to either parental form. For example, in snapdragons, AA results in red flowers while aa yields white ones; Aa produces pinkish-red blooms that blend features of both original colors.
– Dominant-Recessive Inheritance: One allele (A) masks the expression of another (a), leading to a single phenotypic outcome. For instance, brown eyes are dominant over blue eyes in humans.
– Analogously, heterozygosity is akin to possessing two distinct cookbooks. When instructions for preparing a dish differ between books – say, one calls for more sugar while the other uses less salt – combining these methods might result in an intermediate or unique outcome that blends aspects of both original recipes.
Understanding genotype classification enables researchers and breeders alike to predict phenotypic outcomes based on genetic constitutions, facilitating studies into inheritance patterns, trait mapping, and selective breeding programs.
Genetic Diversity (simple definition): The variety of genes within a population enables evolution and adaptation. Genetic diversity encompasses the myriad genes present within a population or species, serving as the raw material for evolution and adaptation.
Genetic Load: Harmful mutations that negatively impact fitness accumulate over generations. Conversely, genetic load refers to the accumulation of deleterious mutations that can negatively impact an organism’s fitness, robustness, and reproductive success.
Adaptation: On one hand, introducing beneficial genetic diversity can enhance adaptability and evolutionary potential by providing a broader gene pool from which advantageous traits may emerge or be recombined. However, this process must be navigated judiciously to avoid inadvertently increasing the detrimental effects of genetic load – the presence of harmful alleles that could negatively impact fitness if expressed. Outbreeding risks include: 1. Outbreeding Depression: Introducing too many novel alleles may disrupt locally adapted gene complexes, reducing offspring fitness. 2. Migration Load: New alleles might carry hidden deleterious mutations that become expressed under changed conditions, potentially harming population viability.